医疗器械的备案与注册
- 供应商
- 南方泰利(厦门)信息科技有限公司
- 认证
- 联系电话
- 13306003311
- 手机号
- 13306003311
- 总经理
- 邓瑞灿
- 所在地
- 厦门思明区银河大厦19E
- 更新时间
- 2023-10-31 13:09
1、为什么要进行备案和注册?
2014年版《医疗器械监督管理条例》第八条的规定,国家对第二类、第三类医疗器械实行注册管理,对第一类医疗器械实行备案管理。医疗器械注册人、备案人以自己名义把产品推向市场,对产品负法律责任。
至于医疗器械分类,可以参考之前的文章。
数据之路:医疗器械简介7赞同 · 3 评论文章
2、什么是医疗器械的备案与注册
医疗器械备案:指医疗器械备案人向药品监督管理部门提交备案资料,药品监督管理部门对提交的备案资料存档备查。
医疗器械注册:指药品监督管理部门根据医疗器械注册申请人的申请,依照法定程序,对其拟上市医疗器械的安全性、有效性研究及其结果进行系统评价,以决定是否同意其申请的过程。
3、备案注册的实践发展
(1)1996年,原国家医药管理局发布《医疗器械注册管理办法》
(2)2004年,原国家食品药品监督管理局修订《医疗器械注册管理办法》
(3)2007年,原国家食品药品监督管理局颁布《体外诊断试剂注册管理办法》
(4)2014年,国家实施新的《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》
4、医疗器械备案资料及对应机关
4.1医疗器械产品备案递交资料

(1)产品技术要求
产品技术要求主要包括医疗器械成品的性能指标和检验方法。产品技术要求应该按照《医疗器械产品技术要求编写指导原则》编制。
(2)产品检验报告
产品检验报告可以是备案人的自检报告。
(3)临床评价资料
临床评价资料不一定是临床试验报告,当然有临床试验报告更好,更能满足要求。可以是通过文献、同类产品临床使用获得的数据证明该医疗器械安全、有效的资料,即同品种临床评价资料。
(4)产品说明书及标签样稿
(5)与产品研制、生产有关的质量管理体系文件
(6)证明产品安全、有效所需的其他资料
(7)产品风险分析资料
应按照yy/t0316《医疗器械风险管理对医疗器械的应用》有关要求编制按安全风险分析报告,主要包括:医疗器械预期用途和安全性有关特征的判定,对每个已判定的危害处境,评价和决定是否需要降低风险,风险控制措施的实施和验证结果,必要是引用检测和评价性报告,任何一个或多个剩余风险的可接受性评定等内容。
(8)第一类医疗器械备案信息表

4.2备案机关
按照《医疗器械监督管理条例》,境内第一类医疗器械,在市级药品监督管理机构备案。境外第一类医疗器械,在国家药品监督管理局备案,港澳台参考境外医疗器械办理。

4.3备案流程
4.3.1第一类医疗器械备案号
第一类医疗器械备案号的编排方式为:×1械备××××2××××3号。
×1为备案部门所在地的简称:
进口第一类医疗器械为“国”字;
境内第一类医疗器械为备案部门所在的省、自治区、直辖市简称加所在设区的市级行政区域的简称(无相应设区的市级行政区域时,仅为省、自治区、直辖市的简称);
××××2为备案年份;
××××3为备案流水号。
4.3.2备案变更
(1)内容变更
已备案的医疗器械,备案信息表中登载内容及备案的产品技术要求发生变化的,备案人应当提交变化情况的说明及相关证明文件,向原备案部门提出变更备案信息。
备案资料符合形式要求的,药品监督管理部门应当将变更情况登载于变更信息中,将备案资料存档。
(2)管理类别变更
备案的医疗器械管理类别调整的,备案人应当主动向药品监督管理部门提出取消原备案;管理类别调整为第二类或者第三类医疗器械的,按照《医疗器械注册管理办法》规定申请注册,也就是从备案升级为注册。
(3)变更备案资料
l变化情况说明及相关证明文件
证明性文件
符合性声明
5、医疗器械注册资料及对应机关
5.1注册申请
申请医疗器械注册,申请人应当按照相关要求向药品监督管理部门报送申报资料。
5.2医疗器械注册申报资料要求及注册机关
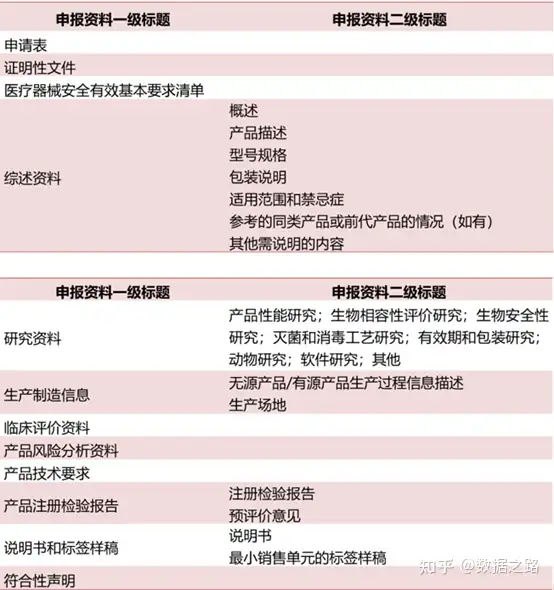
1、申请表
(1)境内申请人提交:企业营业执照副本复印件和组织机构代码证复印件。按照《创新医疗器械特别审评程序审批》的境内医疗器械申请注册时,应当提交创新医疗器械特别审批申请审查通知单。
(2)境外申请人应当提交:a.境外申请人注册地或生产地地址所在国家(地区)医疗器械主管部门出具的允许产品上市销售的证明文件、企业资格证明文件;b.境外申请人在中国境内指定代理人的委托书、代理人承诺书及营业执照副本复印件或机构登记证明复印件。
2、生产制造信息
应当明确产品生产加工工艺,明确关键工序和特殊过程。
3、产品注册检验报告
提供具有医疗器械检验资质的医疗器械检验机构出具的注册检验报告和预评价意见。